We investigate different approaches for the classification of proteins based on sequence data, employing supervised machine learning techniques. Among many other methods for protein classification, multiple sequence alignment of proteins within a known family has been a popular way to extract statistical information about a protein family that would not otherwise be observed to the human eye. Profile hidden Markov models (HMMs) are then built from multiple alignment of raw sequence data and random forests (RFs) are used to discriminate between two sets of proteins based on features (e.g., functional amino acid groups properties extracted from the raw sequences).
HMM shows that sequence alignment approaches determine small differences between similar proteins well, while RF specializes in classifying proteins that do not share high sequence homology within the family as biological features are the basis for classification. Compared to artificial neural networks (ANNs) and support vector machine (SVM), the machine learning based methods achieve a higher classification accuracy.
 |
| The two prion protein nucleotide sequences are identical apart from one element in the sequence. How long does it take to spot this by eye? |
Protein Classification - Randomized Decision Trees
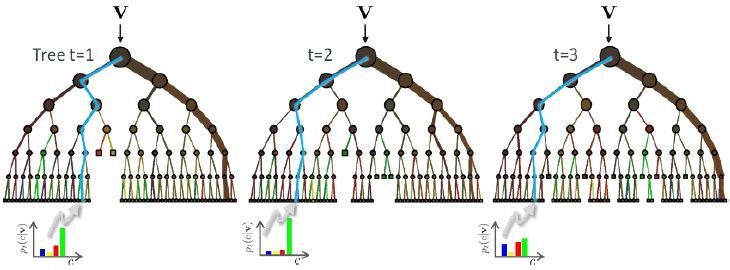 |
| All test data moving through each tree reaches a leaf node based on the optimization of the parameters held at each node. The result of the leaf node is a weak learner in which the average of all trees are taken as the classification prediction. |
 |
| Protein features. |
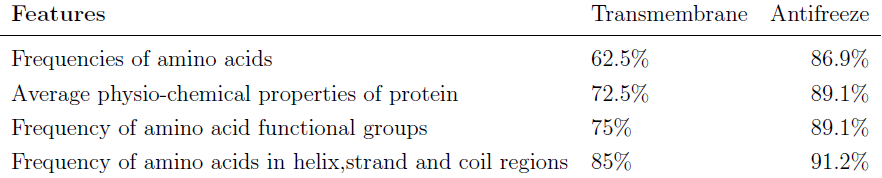 |
| Cumulative classification accuracy rates. |
Protein Classification - Profile Hidden Markov Models
| Attach:hmm.png Δ |
| An example of a profile HMM. States B and E are beginning and end states. Match states are a normal part of both aligned and unaligned sequences, but delete/insert states are needed to create a p-HMM based on aligned data. Delete states do not emit anything from the model, and insert states can emit any amino acid, and can either transition back to itself, or to a match state. |
 |
| The training process of each HMM model. |
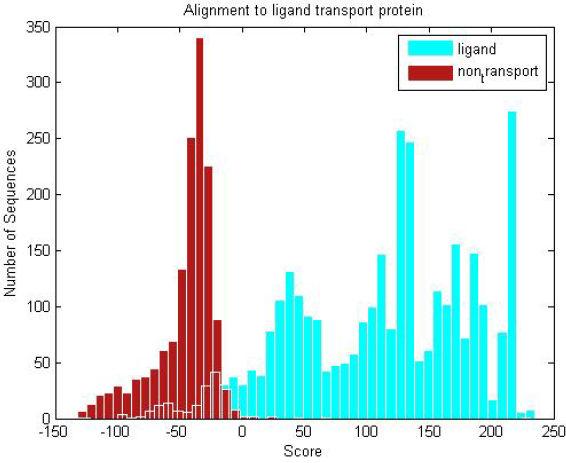 |
| Ligand transmembranes and non-transmembrane proteins are scored against the ligand profile HMM. The distribution clearly shows the trained ligand HMM can discriminate between the two groups of proteins. |
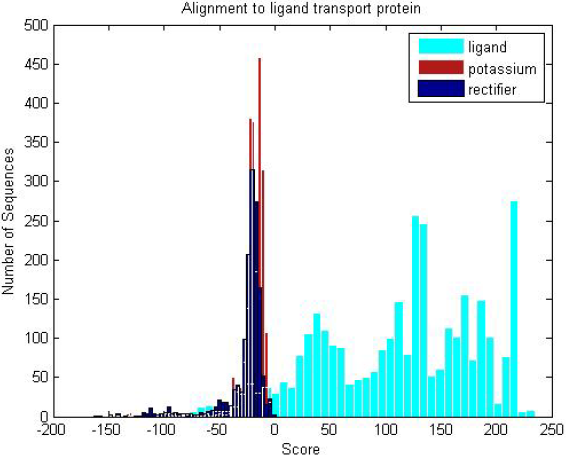 |
| Three transmembrane sub-groups (ligand,potassium and recti�er) are scored against the ligand profile HMM, where the ligand HMM scores significantly higher for ligand proteins than the other transmembranes, despite stark similarities in structure. |
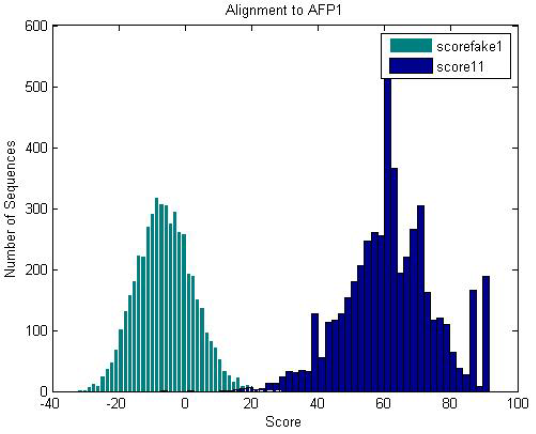 |
| Fake proteins generated with the same amino acid frequency as the antifreeze proteins scored against the antifreeze HMM is significantly less than that of the antifreeze proteins. |
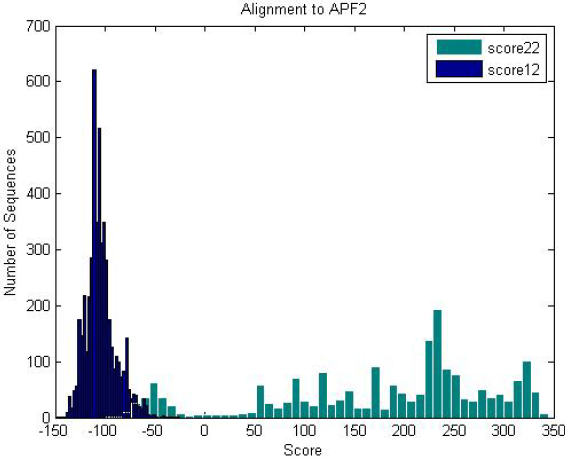 |
| Antifreeze-like (green) and antifreeze proteins (blue) scored against an antifreeze-like HMM. Antifreeze proteins do not score well because antifreeze-like proteins have high variance in their sequence data, providing various functional properties. |
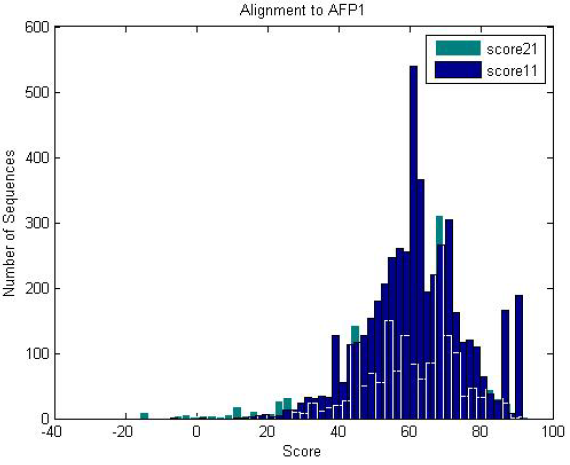 |
| Antifreeze-like (green) and antifreeze proteins (blue) scored against an antifreeze HMM. Both of them score well against the HMM, where antifreeze-like proteins contain segments of sequence similarity to antifreeze proteins. |
Publications
- A. Lacey, J. Deng, X. Xie, S.-K. Chung, J. Mullins, and M. I. Rees, Protein Classification using Hidden Markov Models and Random Forests, In Proc. Int'l Conf. BioMedical Engineering and Informatics, October 2014.
- A. Lacey, Supervised Machine Learning Techniques in Bioinformatics: Protein Classification, MSc by Research Thesis, Swansea University, May 2014.

{kind=link}